Feedback
For methodology and benchmark queries, please refer to the manuscript in PLOS ONE.
For technical or results queries, please contact: gut-dock|chem.uw.edu.pl
For formal or terms of use queries, please contact: Laboratory of Modeling of Cellular Processes.
Description
GUT-DOCK is a web service for prediction of binding affinities of small-molecule ligands for gut hormone G protein-coupled receptors. GUT-DOCK incorporates Autodock VINA and OpenBabel for ligand preparation and docking. Most of receptors included in GUT-DOCK belong to gut hormone receptors and other class B receptors which are expressed in the gastrointestinal tract. GUT-DOCK performs docking of User's compound to GCGR, GLP-1R, GIPR1, VIPR1, and PAC-1R receptors. All receptors are included in their inactive-state conformations. For more information on gut hormone receptors which are involved in the regulation of glucose and insulin homeostasis please visit, for example, the Glucagon website.
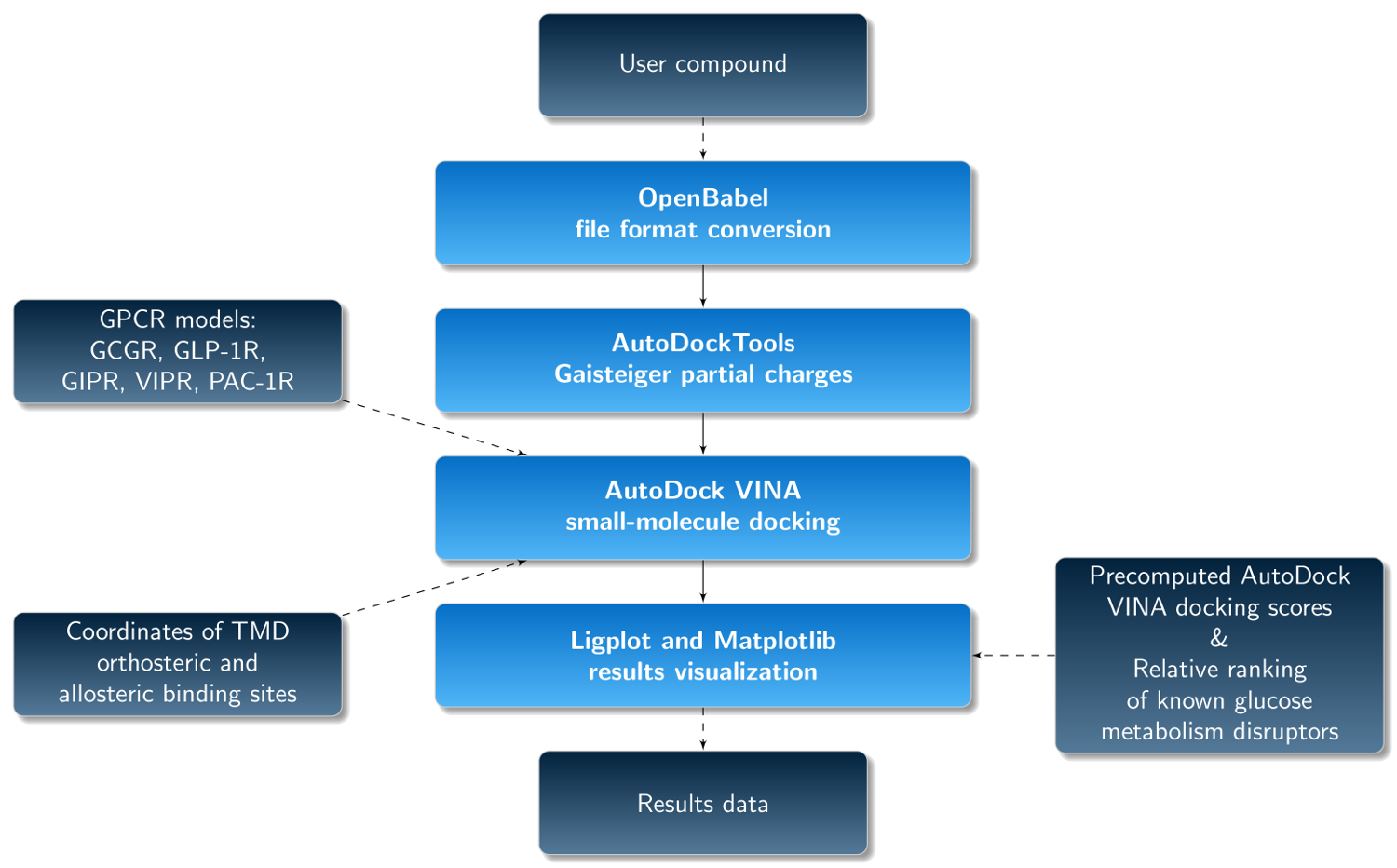
Fig. 1 The pipeline of the GUT-DOCK web service. In the current version, precomputed AutoDock VINA docking scores and relative ranking for known ligands of gut hormone receptors from ChEMBL has been added.
Query form
| User name |
Maximum numbers of characters: 100 |
| E-mail address | Please, enter a valid E-mail address. A link referring to results will be sent via E-mail. Otherwise, the results website can be also saved in bookmarks. |
| Job description |
Maximum numbers of characters: 1000 |
| Ligand binding site | It is required to select either orthosteric or allosteric binding site for docking. To date, in all available crystal structures of class B GPCRs, small molecule ligands (negative allosteric modulators) were reported to be bound to the allosteric site. The orthosteric binding site of class B GPCRs is usually occupied by endogenous peptides. |
| Ligand file |
Allowed file formats: pdbqt, pdbq, pdb, mol2, sdf, smi, and mol. File type is recognized by filename extension. For more reliable results, it is advised to submit files including a 3D structure of a ligand with added hydrogen atoms and respective partial charges. Otherwise, the ligand file is automatically converted with OpenBabel and partial atomic charges are added automatically with AutodockTools. There are rare cases when OpenBabel fails to convert a ligand file into the PDBQT file format. In such cases we advice to convert the failed ligand file into any other format using a standalone program such as, e.g., Maestro, Pymol, VMD. Maximum allowed file size: 10 MB. In principle, also peptides ligands can be docked with Autodock VINA. Yet, due to a significant conformational variability of peptide ligands it is reasonable to dock only short peptides (ca. 5 residues) or peptide fragments. Before docking a peptide ligand User should slightly edit its pdb file like in an example below. In the below example we used a small 5 amino acids-long fragment of glucagon derived from a pdb file of GCGR (PDB id: 5YQZ). Main format differences are as follows: HETATM fields instead of original ATOM fields, LIG fields instead of 3-letter codes of amino acids, 1-letter symbols of atoms instead of detailed information (e.g. CA, CB, etc.). Notably, an input conformation of a peptide ligand can be changed because GUT-DOCK imploys a flexible-ligand docking procedure. An original pdb file of a peptide:
ATOM 1 N SER 2 -17.641 25.582 9.047 1.00 94.83 N ATOM 2 CA SER 2 -17.865 25.510 10.486 1.00 93.56 C ATOM 3 C SER 2 -17.799 24.078 11.002 1.00 95.71 C ATOM 4 O SER 2 -17.815 23.886 12.218 1.00 96.23 O ATOM 5 CB SER 2 -19.208 26.132 10.846 1.00 96.76 C ATOM 6 OG SER 2 -19.215 27.517 10.548 1.00106.48 O ATOM 7 N GLN 3 -17.707 23.075 10.085 1.00 89.57 N ATOM 8 CA GLN 3 -17.629 21.639 10.403 1.00 87.68 C ATOM 9 C GLN 3 -16.570 21.327 11.458 1.00 88.82 C ATOM 10 O GLN 3 -16.912 20.821 12.532 1.00 87.72 O ATOM 11 CB GLN 3 -17.436 20.791 9.131 1.00 88.54 C ATOM 12 CG GLN 3 -17.470 19.280 9.374 1.00 93.86 C ATOM 13 CD GLN 3 -18.696 18.834 10.133 1.00107.29 C ATOM 14 NE2 GLN 3 -18.506 18.391 11.359 1.00 95.43 N ATOM 15 OE1 GLN 3 -19.820 18.881 9.633 1.00104.16 O ATOM 16 N GLY 4 -15.320 21.682 11.156 1.00 83.96 N ATOM 17 CA GLY 4 -14.196 21.534 12.068 1.00 83.79 C ATOM 18 C GLY 4 -14.415 22.178 13.425 1.00 87.90 C ATOM 19 O GLY 4 -14.060 21.589 14.448 1.00 87.98 O ATOM 20 N THR 5 -15.025 23.383 13.458 1.00 85.05 N ATOM 21 CA THR 5 -15.353 24.170 14.662 1.00 83.71 C ATOM 22 C THR 5 -16.486 23.482 15.477 1.00 89.82 C ATOM 23 O THR 5 -16.498 23.568 16.707 1.00 89.79 O ATOM 24 CB THR 5 -15.687 25.616 14.264 1.00 87.75 C ATOM 25 CG2 THR 5 -15.640 26.577 15.435 1.00 80.09 C ATOM 26 OG1 THR 5 -14.788 26.052 13.243 1.00 94.07 O ATOM 27 N PHE 6 -17.449 22.824 14.791 1.00 85.58 N ATOM 28 CA PHE 6 -18.536 22.114 15.464 1.00 83.64 C ATOM 29 C PHE 6 -17.921 20.849 16.062 1.00 85.41 C ATOM 30 O PHE 6 -17.890 20.730 17.290 1.00 84.57 O ATOM 31 CB PHE 6 -19.657 21.799 14.474 1.00 84.49 C ATOM 32 CG PHE 6 -20.897 21.132 15.008 1.00 84.59 C ATOM 33 CD1 PHE 6 -22.044 21.868 15.266 1.00 86.87 C ATOM 34 CD2 PHE 6 -20.946 19.753 15.178 1.00 85.68 C ATOM 35 CE1 PHE 6 -23.206 21.242 15.745 1.00 88.26 C ATOM 36 CE2 PHE 6 -22.108 19.125 15.640 1.00 89.34 C ATOM 37 CZ PHE 6 -23.231 19.873 15.922 1.00 87.87 C An edited pdb file of a peptide that fits GUT-DOCK requirements:
HETATM 1 N LIG 1 -17.641 25.582 9.047 1.00 94.83 N HETATM 2 C LIG 1 -17.865 25.510 10.486 1.00 93.56 C HETATM 3 C LIG 1 -17.799 24.078 11.002 1.00 95.71 C HETATM 4 O LIG 1 -17.815 23.886 12.218 1.00 96.23 O HETATM 5 C LIG 1 -19.208 26.132 10.846 1.00 96.76 C HETATM 6 O LIG 1 -19.215 27.517 10.548 1.00106.48 O HETATM 7 N LIG 1 -17.707 23.075 10.085 1.00 89.57 N HETATM 8 C LIG 1 -17.629 21.639 10.403 1.00 87.68 C HETATM 9 C LIG 1 -16.570 21.327 11.458 1.00 88.82 C HETATM 10 O LIG 1 -16.912 20.821 12.532 1.00 87.72 O HETATM 11 C LIG 1 -17.436 20.791 9.131 1.00 88.54 C HETATM 12 C LIG 1 -17.470 19.280 9.374 1.00 93.86 C HETATM 13 C LIG 1 -18.696 18.834 10.133 1.00107.29 C HETATM 14 N LIG 1 -18.506 18.391 11.359 1.00 95.43 N HETATM 15 O LIG 1 -19.820 18.881 9.633 1.00104.16 O HETATM 16 N LIG 1 -15.320 21.682 11.156 1.00 83.96 N HETATM 17 C LIG 1 -14.196 21.534 12.068 1.00 83.79 C HETATM 18 C LIG 1 -14.415 22.178 13.425 1.00 87.90 C HETATM 19 O LIG 1 -14.060 21.589 14.448 1.00 87.98 O HETATM 20 N LIG 1 -15.025 23.383 13.458 1.00 85.05 N HETATM 21 C LIG 1 -15.353 24.170 14.662 1.00 83.71 C HETATM 22 C LIG 1 -16.486 23.482 15.477 1.00 89.82 C HETATM 23 O LIG 1 -16.498 23.568 16.707 1.00 89.79 O HETATM 24 C LIG 1 -15.687 25.616 14.264 1.00 87.75 C HETATM 25 C LIG 1 -15.640 26.577 15.435 1.00 80.09 C HETATM 26 O LIG 1 -14.788 26.052 13.243 1.00 94.07 O HETATM 27 N LIG 1 -17.449 22.824 14.791 1.00 85.58 N HETATM 28 C LIG 1 -18.536 22.114 15.464 1.00 83.64 C HETATM 29 C LIG 1 -17.921 20.849 16.062 1.00 85.41 C HETATM 30 O LIG 1 -17.890 20.730 17.290 1.00 84.57 O HETATM 31 C LIG 1 -19.657 21.799 14.474 1.00 84.49 C HETATM 32 C LIG 1 -20.897 21.132 15.008 1.00 84.59 C HETATM 33 C LIG 1 -22.044 21.868 15.266 1.00 86.87 C HETATM 34 C LIG 1 -20.946 19.753 15.178 1.00 85.68 C HETATM 35 C LIG 1 -23.206 21.242 15.745 1.00 88.26 C HETATM 36 C LIG 1 -22.108 19.125 15.640 1.00 89.34 C HETATM 37 C LIG 1 -23.231 19.873 15.922 1.00 87.87 C |
Results
As a result GUT-DOCK provides possible ligand binding modes for several GPCRs. 3D coordinates of ligand-receptor complexes can be downloaded as PDB files. Ligand interactions are visualized with Ligplot. Additionaly, predicted ligand binding affinity for a given receptor is provided, based on Autodock VINA scoring function. More details on GUT-DOCK are provided in references.
Predicted ligand binding affinity based on Autodock VINA for each GPCR receptor is juxtaposed with VINA-precomputed binding affinities of known active ligands retrieved from ChEMBL. Here, 6 ranks corresponding to 6 ranges of ligand activity based on pChEMBL values were assigned to each ligand (Dragan et al. IJMS 2023). Rank 6: 0.0 - 5.0; rank 5: 5.0 - 6.0; rank 4: 6.0 - 7.0; rank 3: 7.0 - 8.0; rank 2: 8.0 - 9.0; rank 1: pChEMBL above 9.0 (see Table 1).
Table 1. Autodock VINA results for known GCGR negative allosteric modulators vs. their relative ranks based on pChEMBL values.
| ChEMBL ID | 2D Structure | pChEMBL values | Rank based on pChEMBL values (best for highest) | Autodock VINA results for allosteric site of GCGR (NAMs binding site) |
|---|---|---|---|---|
| CHEMBL487476 |  |
8.41 | 2 | -8.9 |
| CHEMBL455214 |  |
8.22 | 2 | -9.5 |
| CHEMBL1922696 |  |
7.64 | 3 | -8.8 |
| CHEMBL452310 | 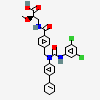 |
6.16 | 4 | -7.9 |
| CHEMBL441160 | 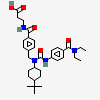 |
5.62 | 5 | -7.4 |
Additionally, known glucose homeostasis disruptors among commonly used drugs (Latek et al. 2019) were added with rank equal to 6. In the previous version of GUT-DOCK (Pasznik et al. 2019) only beta-blockers were included for comparison of Autodock VINA-predicted binding affinities (see Table 2).
Table 2. Autodock VINA results for beta-blockers vs. their relative disrupting effect on glucose homeostasis (Pasznik et al. 2019).
Name Rank based on Autodock VINA results for clinical trials allosteric site of GCGR (best for diabetics) (NAMs binding site) nebivolol 1 -8.1 carvedilol 1 -7.0 labetalol 2 -7.2 atenolol 3 -5.8 metoprolol 4 -5.5
In the previous version of GUT-DOCK, the above ranks refered to the probability of inducing diabetes during pharmacotherapy (e.g., a number of patients who developed new-onset diabetes during treatment). However, clinical data for drug-induced diabetes is often contradictory (see References). What is more, the molecular mechanism of action of novel beta-blockers associated with better prognosis for diabetics has not been described so far. In the current version of GUT-DOCK (Latek et al. in preparation) mostly results from binding and functional assays has been included instead of clinical data. Thus, the predicted binding affinity of User's compound for a certain receptor type can be juxtaposed with predicted binding affinities of known active ligands for this receptor.
Standard settings for Ligplot are used for visualization of polar contacts and hydrogen bonds. By comparing values of binding affinities approximated by docking scores it is possible to predict the best protein target for a submitted compound among available class B GPCRs. Accuracy of such approach is limited by performance of Autodock VINA in virtual screening tasks.